Sickle cell anaemia

Professor John Gribben - Haematology
Created on: 01-08-2018
Updated on: 10-03-2023
Edited by: Carlota Pano
What is sickle cell anaemia?
Sickle cell anaemia is the most well-known and common type of sickle cell disease, in which the blood disorder results in anaemia (a shortage of haemoglobin and/or red blood cells in the blood, reducing the amount of oxygen carried by the blood to the cells that need it).
Haemoglobin is a protein found in red blood cells that binds to oxygen, allowing them to carry it around the body to cells that need it to function and grow.
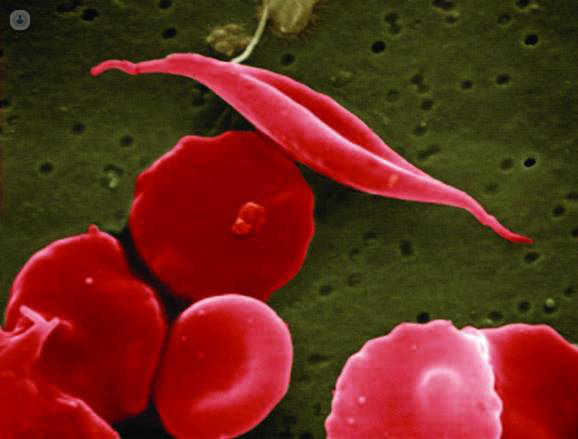
In sickle cell anaemia, haemoglobin forms stiff rods inside the red blood cells, making them rigid and inflexible. While red blood cells are usually round, flexible donut-like structures that pass easily through blood vessels, sickle cells are curved and inflexible due to the rigid haemoglobin, and frequently get stuck, clogging blood vessels and causing pain and damage.
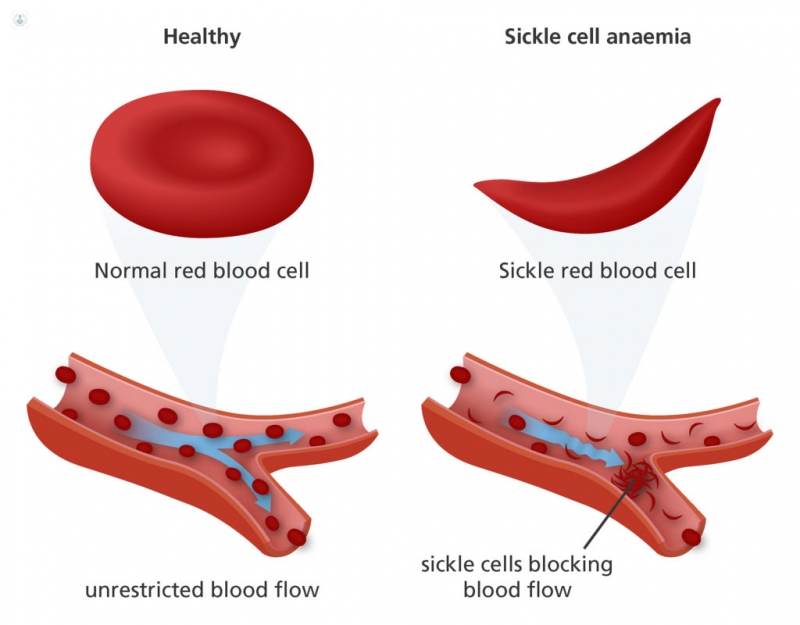
What are the symptoms of sickle cell anaemia?
While red blood cells are usually replaced every 120 days, sickle cells die much faster, typically lasting 10-20 days, causing a shortage of red blood cells, and therefore, a shortage of oxygen to the rest of the body.
This causes classic symptoms of anaemia, such as:
- shortness of breath
- tiredness
- feeling weak
- dizziness
- headaches
Other symptoms of sickle cell anaemia include:
- Cold extremities
- Painful swelling of the hands and feet
- Episodes of pain – the misshapen red blood cells can clog blood vessels, causing intense pain crises. While some people may only suffer a few of such episodes, others have more than a dozen crises a year. Some crises are so serious that the patient is hospitalised.
- Chronic pain – in some people, the condition can cause damage to the patient’s bones, joints, and other parts of the body, or can cause ulcers. These effects can cause long-term pain.
- Frequent infections – if the sickle cells damage an organ like the spleen, which helps the immune system, the patient can become more prone to infection.
- Vision problems – this can occur if the sickle cells block the tiny blood vessels that supply the retina.
- Delayed growth – infants and children need oxygen and nutrients carried by red blood cells to grow, and the lack of this can slow growth.
What causes sickle cell anaemia?
Sickle cell anaemia is a genetic condition. It is caused by a mutation in the gene that codes for haemoglobin. We all inherit two copies of each gene – one from each parent. Sickle cell anaemia is recessive, which means that both of the patient’s copies of the haemoglobin gene must be the mutated form known as “haemoglobin S” for the condition to manifest (meaning that both parents either had the condition themselves, or were carriers).
People who have one normal and one mutated copy of the gene will have sickle cell trait, which means they produce both normal and sickle cell haemoglobin. Their blood may contain a combination of normal red blood cells and sickle cells, but symptoms don’t usually manifest. However, they carry the possibility of passing it on to their children.
What is the treatment?
Treatment is generally geared towards managing pain crises and preventing infection, and any other complications caused by sickle cell anaemia. It is important for the patient to be monitored, with regular eye exams and tests for pulmonary hypertension.
The only “cure” is a bone marrow transplant, which involves destroying the patient’s stem cells and replacing them with those of a donor. Stem cells can develop into any cell as required by the body, and in theory, stem cells from a donor who doesn’t have sickle cell anaemia should develop into healthy, normal red blood cells. However, donors must be blood relatives, and even then, there is a risk that the patient’s body will reject the donated stem cells. The risk of complications is greater the older the patient gets, and for this reason this treatment is performed more frequently on children than adults. Even so, with the shortage of viable donors and the risks involved, the procedure is relatively uncommon.
Experts in Sickle cell anaemia
-
Professor John Gribben
HaematologyExpert in:
- Leukaemia
- Lymphoma
- Stem cells
- Multiple myeloma
- Sickle cell anaemia
- See all

London Haematology
London Haematology
22 Devonshire Place, W1G 6JA
No existe teléfono en el centro.
By using the telephone number provided by TOP DOCTORS, you automatically agree to let us use your phone number for statistical and commercial purposes. For further information, read our Privacy Policy
Top Doctors
-

London Haematology
22 Devonshire Place, W1G 6JA, W1G Marylebone LondonExpert in:
- CAR-T therapy
- Haemato-oncology
- Haematology
- Myeloproliferative disorders
- Red blood cell disorders
- Stem cell transplant
- Most viewed diseases, medical tests, and treatments
- Thrombophilia
- Bowel cancer
- Liver cancer
- Chronic fatigue
- Thalassaemia
- Lymphoma
- Hodgkin's disease
- Iron metabolism disorders
- Stomach cancer
- Haemochromatosis