Sickle cell disease

Dr Nnenna Osuji - Haematology
Created on: 11-22-2017
Updated on: 09-19-2023
Edited by: Kate Forristal
What is sickle cell disease?
Sickle cell disease (SCD) is a group of blood disorders affecting the haemoglobin molecules in red blood cells.
Haemoglobin is a protein found in all red blood cells. It binds to oxygen when the blood passes through the lungs, allowing the red blood cells to carry the oxygen throughout the body.
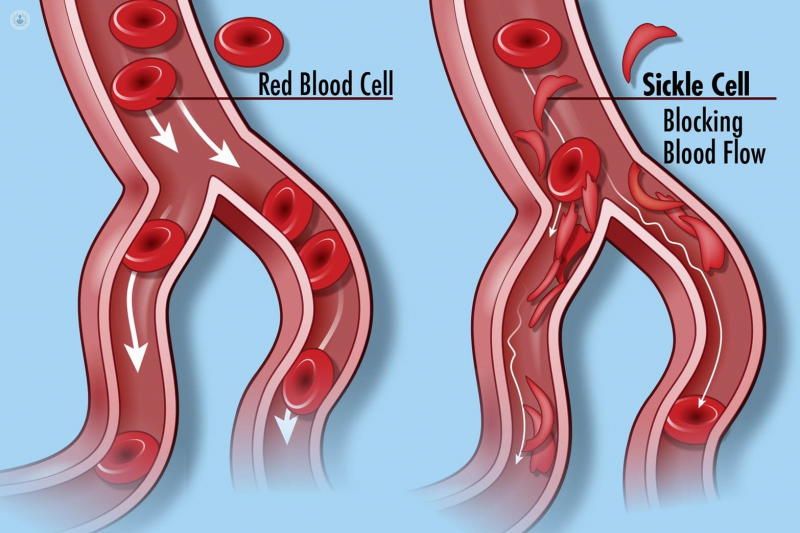
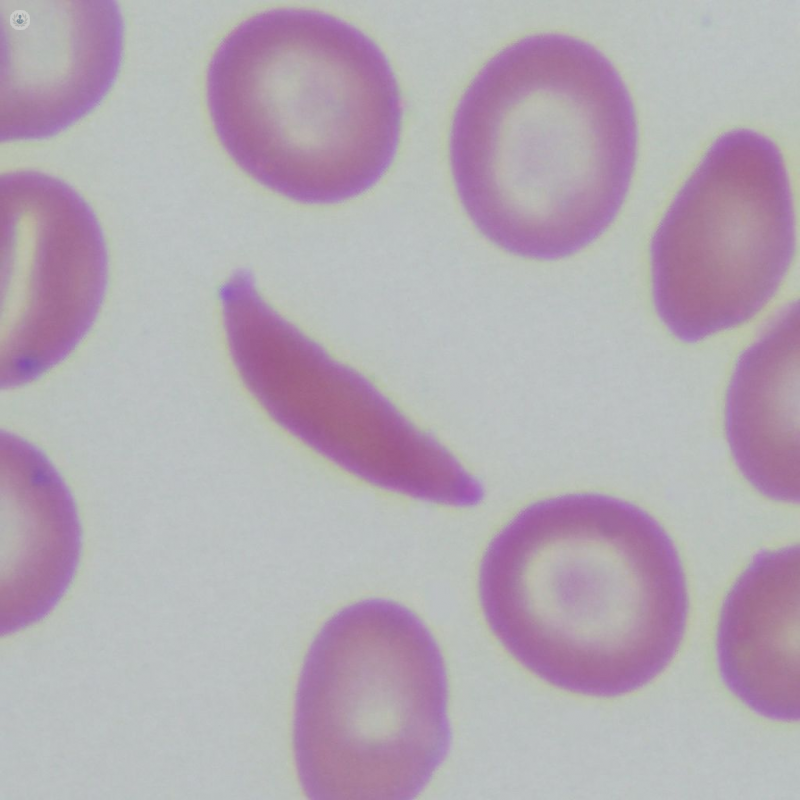
When haemoglobin is abnormal, as in sickle cell disease, it affects its ability to carry oxygen, and can cause the red blood cells to become curved and rigid. This gives rise to the “sickle” shape of the cells.
The most common and most serious type of sickle cell disease is when a person has two haemoglobin “S” genes, and is widely known as sickle cell anaemia. Other common types include haemoglobin SC disease and haemoglobin Sβ thalassaemia.
What are the symptoms?
Sickle cell disease can cause anaemia (low red blood cell count), repeated infections, shortness of breath, fatigue, and episodes of pain called “sickle cell crises”. While a sickle cell crisis might be mild in one patient, in most they are intense, and may require treatment at a hospital.
Some patients may experience chronic (long-term) pain, while children with the condition may have poor growth or delayed development. Over time, sickle cell disease can cause organ damage, including to the brain, kidneys, liver, lungs, eyes, heart, spleen, genitals, joints, and skin.
What causes sickle cell disease?
Sickle cell disease is genetic, caused by a mutation on chromosome 11. We each inherit two copies of each gene (one from each parent). When a person is born with two abnormal haemoglobin genes, one of which being haemoglobin S, they will have some form of SCD. If both copies are S, the form that manifests will be sickle cell anaemia.
The abnormal haemoglobin changes the make-up of red blood cells. Normally, red blood cells are shaped like a donut without the hole. These smooth, disc-like structures filled with normal haemoglobin are flexible, and pass easily through small blood vessels, delivering vital oxygen to the body’s tissues. Abnormal haemoglobin forms stiff rods within cells, hampering their flexibility and manoeuvrability.
In the more severe forms, like sickle cell anaemia, the red blood cells become awkward, curved (“sickle-shaped”) structures. In addition to not being able to carry oxygen as efficiently, they can clog small blood vessels, preventing oxygen from reaching the tissues, causing sudden pain crises, and over time, organ damage.
When the cells are unable to move freely, they burst (haemolyse), which forces the body to make more. If it is unable to keep up with the demand for red blood cells, the patient becomes anaemic, and has less energy.
How can it be prevented?
There is no way to prevent sickle cell disease, as it is genetic, and a person is either born with it or without it. However, it is possible to test for the disease via a blood test, including during pregnancy. This allows the patient to take steps to manage the illness and improve quality of life.
What is the treatment?
The only known cure is a bone marrow transplant, in which the patient’s natural stem cells in their bone marrow are reduced or destroyed, and donated stem cells (from a donor without sickle cell disease) are injected, and will find their way to the recipient’s bone marrow and multiply. Stem cells can develop into any kind of cells, including red blood cells, and in theory, the new stem cells should create normal, functioning red blood cells. However, the donor has to be a close match, such as a brother or sister, and even then, there is a danger that the recipient’s body will reject the injected stem cells. Due to the lack of acceptable donors, this treatment is uncommon, and is used more frequently on children than adults, as the procedure becomes more risky with age.
Living with sickle cell disease means having treatment to reduce or prevent complications, such as taking penicillin and keeping up to date on immunisations to prevent infections (which can be serious if, as in many cases, the spleen isn’t functioning properly). Careful monitoring of the patient’s health by doctors is essential, including regular eye exams and tests for pulmonary hypertension. Pain management may be recommended for sickle cell crises, and sometimes blood transfusions may be necessary.
Experts in Sickle cell disease
-

Professor John Porter
HaematologyExpert in:
- Haemochromatosis
- Anaemia
- Thalassaemia
- Sickle cell disease
- Hyperferritinaemia
- Iron deficiency
-
Dr Nnenna Osuji
HaematologyExpert in:
- Anaemia
- Blood clots
- Sickle cell disease
- Lymphoma
- Leukaemia
- Thrombocytopenia
- See all

Shirley Oaks Hospital - part of Circle Health Group
Shirley Oaks Hospital - part of Circle Health Group
Poppy Lane, Croydon CR9 8AB
No existe teléfono en el centro.
By using the telephone number provided by TOP DOCTORS, you automatically agree to let us use your phone number for statistical and commercial purposes. For further information, read our Privacy Policy
Top Doctors
HCA UK at University College Hospital
HCA UK at University College Hospital
1 Grafton Way
No existe teléfono en el centro.
By using the telephone number provided by TOP DOCTORS, you automatically agree to let us use your phone number for statistical and commercial purposes. For further information, read our Privacy Policy
Top Doctors
-

Shirley Oaks Hospital - part of Circle Health Group
Poppy Lane, Croydon CR9 8AB, South LondonExpert in:
- General Surgery
- Orthopaedic surgery
- Plastic surgery, reconstructive and aesthetics
- Gastroenterology
- Obstetrics and Gynaecology
- Neurosurgery
-

HCA UK at University College Hospital
1 Grafton Way, Central LondonExpert in:
- Stem cells
- Clinical trials
- Haemato-oncology
- Haematology
- Medical Oncology
- Radiotherapy
- Most viewed diseases, medical tests, and treatments
- Thrombophilia
- Bowel cancer
- Liver cancer
- Chronic fatigue
- Thalassaemia
- Lymphoma
- Hodgkin's disease
- Iron metabolism disorders
- Stomach cancer
- Haemochromatosis