Amyotrophic lateral sclerosis (ALS)

Dr Margareta Griesz-Brisson - Neurology
Created on: 01-16-2013
Updated on: 05-02-2023
Edited by: Conor Lynch
What is amyotrophic lateral sclerosis?
Amyotrophic lateral sclerosis (ALS) is a degenerative disease. It is a disease of neurones in the brain, the brain stem, and the spinal cord that control the movement of voluntary muscles, leading to disability.
ALS attacks the motor neurones, which are the longest cells in the body connecting electric signals from the brain to muscles. The cells do not receive proper nutrition and necessities to stay healthy due to the disease and start to deteriorate, eventually leading to cell death. For a full-detailed scientific explanation on the mechanics of ALS, see the official ALS website.
ALS affects approximately 5 in every 100,000 people in the world. It is also known as motor neurone disease (MND) or Lou Gehrig's disease in the U.S..
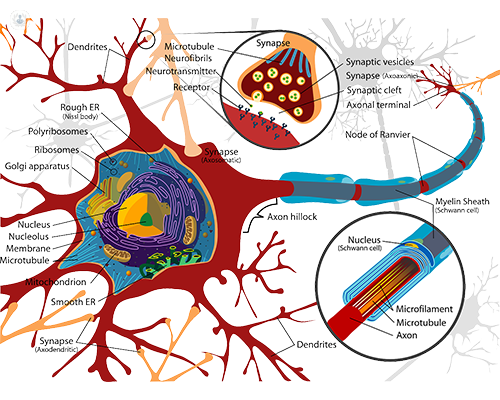
protecting the axon is next, followed by the axon terminal which connects to the muscle. Once all
these parts have deteriorated, the cell will die and no longer be able to send signals to the muscle.
How many variants are there?
There are two known different types of ALS.
- Sporadic ALS: this is the most common type of ALS where it can affect anyone. It makes up approximately 90 to 95 per cent of all ALS cases.
- Familial (Genetic) ALS: this type is passed down genetically. If a family has genetic ALS, the chances of each child having it will be 50 per cent. This type makes up approximately 5 to 10 per cent of all cases.
Within each type, there are two different classifications for onset.
- Spinal/Limb-onset ALS: making up approximately 60 per cent of all cases, spinal ALS appears as weakness in the arms, legs, and similar areas. It is a rather swift-acting form with death occurring within three to five years.
- Bulbar ALS: this less common form first appears as speech and swallowing difficulty, something that appears later in spinal ALS. Bulbar ALS tends to affect woman more than men and those who are over 70 years old. Typically, full paralysis will occur within one to two years.
Other variations of the disease can depend on age, progression rate, and location.
- Guamanian ALS: this variant only affects people in a specific part of the Pacific in Guam and Trust Territories of the Pacific. The reasons for this isolated type are still unknown, but researchers hypothesise that it is related to a unique genetic component.
- Juvenile ALS: this will first appear as facial spasticity and spastic gait in children or young adults before 25 years old. It is an extremely rare form with a strong genetic component.
- Primary Later Sclerosis: this rare form progresses slowly, allowing those diagnosed with it to maintain more motor function than the other types until it becomes debilitating. The average lifespan of someone living with PLS is 20 years.
What is the prognosis?
The prognosis is poor, as 80 per cent of people diagnosed will see a fatality within five years. Normally, the disease leads to death three to five years after the diagnosis. There are exceptions: it has been calculated that 1 in 4 survive beyond five years from the diagnosis. There are also cases that survive longer, usually with the aid of respiratory and nutritional support.
What are the symptoms?
Symptoms do not usually occur before the age of 50. The most common symptom is loss of muscle strength and coordination, that gradually worsens.
This weakness initially affects legs and arms, and as the disease progresses, other parts of the body are affected. Muscle weakness causes the following symptoms:
- difficulty breathing
- problems climbing stairs, walking or lifting objects
- difficulty swallowing; drooling; nausea
- hoarseness and voice changes
- problems talking, more slowly
- neck muscles can no longer support the weight of the head, which droops
Other symptoms that may occur are depression, muscle rigidity (spasticity), muscle contractions (fasciculations), muscle cramps, and weight loss.
Dementia is rare; the majority of people with the disease can think normally and have intact senses.
What are the first signs of ALS?
As ALS affects all the muscles which have voluntary control, the typical first signs of the disease is muscle weakness and/or stiffness and spasticity. Someone who is having early signs of ALS will have difficulty with routine tasks such as eating, speaking, holding things, generally moving, and sometimes even breathing. The hands, arms, and legs are often the first parts of the body where people experience the initial symptoms.
For those with bulbar onset ALS, the first signs will appear as difficulty with swallowing and speaking.
How is it medically tested and diagnosed?
If any of the above-mentioned symptoms occur, a physical examination will be done to determine whether:
- weakness is present, normally in a specific area
- tongue fasciculations
- muscle spasms or tremor
- rigid or clumsy gait
- stronger or weaker reflexes in the joints
- difficulty controlling crying or laughter
- loss of gag reflex
Often times, a patient will be referred to other specialists before being referred to a neurologist, especially if symptoms first appear minor. As soon as the symptoms have been ruled as neurological, a consultation with a neurologist will be arranged.
Other evaluations may include the following:
- blood tests to rule out other diseases
- assessment of breathing to determine whether the lungs are affected
- check if there is a disease or injury of the neck
- electromyography to determine if there are nerves or muscles not functioning correctly
- check whether there is a family history of the disease
- CT scan or MRI scan of the head
- analyse swallowing
- lumbar puncture

He died in 2018 after living with the disease for 55 years.
What is the life expectancy of someone with ALS?
The average life expectancy of someone who has been diagnosed is two to five years after diagnosis. However, some variations of the disease, such as PLS, can see a life expectancy of up to 20 years. Others may live up to 10 years.
The main cause of fatality amongst those with ALS is respiratory failure, or malnutrition and dehydration. If these two aspects can be managed well and excellent care is provided, there can be an extension of the life expectancy.
Of course, there are some outliers such as Stephen Hawking, who was able to live for 55 years with the disease. This circumstance was a combination of the biology of his type of ALS and the excellent care he received.
What are the causes of ALS?
In most cases, the cause is not known. It has been calculated that one in 10 cases is due to a genetic defect.
Researchers are still studying what the exact causes are, but it is believed that environmental and lifestyle factors play a role in addition to genetics.
Can it be prevented?
The disease cannot be prevented.
However, those who have been diagnosed with ALS can participate in clinical trials to help researchers learn about potential causes and risk factors.
The current known ways to slow onset in elderly patients include:
- eating red, yellow, and orange vegetables, as these contain carotenoids which ward off cognitive decline and neurodegenerative disorders.
- eating lots of greens containing carotene and lutein.
- increasing intake of vitamin E and Omega-3 fatty acids, which reduces oxidative stress on the neurones.
- exercising regularly, even gentle exercise, combined with healthy diet full of antioxidants.
- having genetic testing and identifying symptoms.
- receiving early treatment to slow down the progression of ALS.
Is it contagious?
No, ALS is not a contagious disease.
What are the treatments for ALS?
There is no specific cure for ALS. The disease must be treated by a multidisciplinary team and the aim is to delay symptoms and to improve the patient’s quality of life.
Rehabilitation and physiotherapy may be important for improving muscle function and overall health. A wheelchair and various orthopaedic devices may be necessary to help muscle function.
Some people think that working out the muscles to build strength will help, however, it has the opposite effect. Muscles need to get a little damaged to rebuild stronger in a healthy person, which cannot happen in those with ALS and can actually progress the disease faster.
Does it affect other parts of the brain?
It was previously believed that cognitive function was not affected by ALS. However, recent studies done using MRI scans and other testing are showing new results.
Researchers believe that around 50 per cent of ALS patients experience mild cognitive and behavioural changes. Even more, they are finding that between 5 to 15 per cent of patients will actually experiences changes that are severe enough to result in dementia.
What is it like living with ALS?
The first experience patients describe going through is the loss of control over muscles. It may feel like the body is less steady. Anything that requires fine movement will become more difficult, such as holding a key or using a pen. It can feel like random cramps or suddenly becoming a little clumsy. Speech may become a bit slurred.
Typically, losing control over muscles that allow walking, talking, swallowing, and breathing will start to weaken and there will be no more control. During this time, the body will still be able to: hear, smell, touch, think, taste, and go to the bathroom. Tasks such as showering, getting dressed, eating, etc. will need to be assisted.
As the disease progresses, whether more slowly or quickly, more and more assistance will be needed. Eventually, respiratory care will be required and feeding tube will need to be inserted. Some patients choose to have the feeding tube inserted during the earlier stages when it is not yet necessary as the body is stronger and will heal better.
Which specialist treats it?
ALS should be treated primarily by a neurologist; however many specialists will participate including physiotherapists, speech therapists, and gastroenterologists.


Amyotrophic Lateral Sclerosis / Motor Neurone Disease: FAQ
By Dr Adnan Al-Araji
2024-07-26
Amyotrophic Lateral Sclerosis (ALS, MND) is a challenging and progressive neurodegenerative disease that affects thousands of people worldwide. Dr Adnan Al-Araji explains the disease, including symptoms and treatment. See more
- Most viewed diseases, medical tests, and treatments
- Carpal tunnel syndrome
- Peripheral nerve block
- Minimally invasive spinal surgery
- Back pain
- Spinal surgery
- ADHD
- Long Covid
- Medicolegal
- Specialist rehabilitation
- Vertigo